Ehlers-Danlos syndrom
synonymer

EDS, Ehlers-Danlos-Meekeren syndrom, Van Meekeren syndrom, Fibrodysplasia elastica generalisata, Dermatolysis, Cutis hyperelastica, "gummihud", etc.
Fransk: Laxité articulaire congénitale multiple
Engl .: Danlos 'syndrom, Meekeren-Ehlers-Danlos syndrom, Chernogubovs syndrom, Sacks syndrom, Sack-Barabas syndrom, Van Meekeren syndrom I.
Russisk: Chernogubov syndrom
Definisjon / introduksjon
De Ehlers-Danlos syndrom (EDS) oppsummerer en gruppe heterogen, genetisk Bindevevsykdommer sammen, forårsaket av forstyrrelser i syntesen av kollagen, et strukturelt protein fra Bindevev, betingede og karakteristiske symptomer på hud, leddene og Indre organer utstilling.
Frekvens
De Ehlers-Danlos syndrom er sjelden. Utbredelsen i den totale befolkningen er 1: 5000; 90% av dem er blant dem Type I, II og III påvirket (30% hver), og omtrent 10% av type IV. De andre formene blir sjelden observert.
De Typer I-III bli autosomal dominans arvet, d.v.s. det må bare være et mangelfullt gen for at sykdommen skal bryte ut. De andre gutta vil det autosomal recessivdvs. det må være to defekte gener, eller X-bundetdvs. Sexkromosomoverføring, arvet.
historie
dette ble først beskrevet Ehlers-Danlos syndrom i år 1668 fra Job Janszoon fra Meekeren (1611-1666), kirurg fra Amsterdam. Han hadde oppdaget symptomet på unormal overstretchbarhet hos en spanjol som kunne trekke hakehuden hans opp til øynene og over brystet. Imidlertid observerte han ingen andre avvik.
Først 1891 opprettet hudlegen Chernogubov en fullstendig beskrivelse av det kliniske bildet inkludert ledd- og vaskulær involvering, og det er derfor i russisk medisinsk
Teknisk litteratur til i dag navnet "Chernogubov-syndrom" er vanlig.
Ytterligere beskrivelser fulgte 1901 av den danske hudlegen Eduard Ehlers (1863-1937) og 1908 av hudlegen i Paris Henri A. Danlos (1844-1912). Det var først i 1933 "Ehlers-Danlos Syndrome" som navnet på sykdommen råder.
1949 det var første innsikt i sykdomsfamilien og 1972 var en genetisk feil relatert til det Ehlers-Danlos syndrom oppdaget. I 1986 ble det opprettet en foreløpig klassifisering i 10 typer, som ble endret i 1997 til en forenklet versjon med underinndelingen i seks hovedtyper.
fører til
Årsaken til sykdommen er en Genetisk defekt. Det er en forandring (mutasjon) i genene som utgjør det strukturelle proteinet kollagen beskrive på det menneskelige genomet, DNA. Mutasjonen fører til en endret struktur og / eller en redusert syntese av kollagenet, noe som fører til en redusert styrke av hele bindevevet.
Både Type I og II det er en mutasjon i genkollagen V, der Typer IV en mutasjon i Kollagen III.
symptomer
På grunn av den forstyrrede og reduserte kollagensyntesen, Ehlers-Danlos syndrom de delene av kroppen som er spesielt rik på bindevev: hud, leddene og Blodårer. Siden bindevevet mangler styrke, er det uttrekkbart og rives veldig raskt, noe som er spesielt sant for blodårer som er for små, men noen ganger også massiv blødning kan lede. En viktig komplikasjon er dannelsen av bukker i karene, såkalt. aneurismer, med fare for brudd.
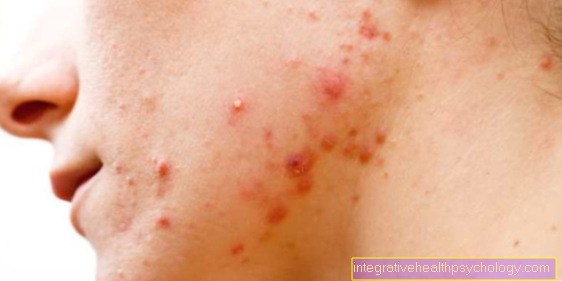
De Hovedsymptom av huden er den mest uttalte Hyperelastisk kuttpå siden av nakke, over leddene og også i ansikt kan løftes opp til 4 cm eller mer. Etter å ha sluppet, springer den umiddelbart tilbake til startposisjonen, og det er grunnen til at den fikk navnet "Gummihud" iført. Generelt sett er huden merkbar tynn (som sigarettpapir), myk og fløyelsaktig ("Marshmallow Skin").
Sår viser forsinket sårheling, slik at suturer tar 3 til 4 ganger lengre tid å leges. Atrofiske eller hypertrofiske, underordnede utvikler seg ofte fra sømmene arr. I tillegg er det dannelse av væskefylte (saftige) hudutbuktninger (mollusoid pseudotumorer) på sterkt belastede områder av kroppen, som f.eks. Kne- og albueleddet, til dannelse av ankelputer ("Knokkelputer") på Baksiden av hånden og foten og av knuter på hæl.
De Skjøter kan bli overstrukket (Hyperflexibility), ofte bevegelig i uønskede retninger og mangler styrke på grunn av løsne leddbånd (Ligament slapphet). Dette kan forårsake uvanlige bevegelser, som de man kan forvente av "bra!" vet. Skjøtene har en tendens til contortions (dislokasjoner) og feilstillinger. De er spesielt berørt skulder- og Ankelledd, den Kneskål (patella), den Temporomandibular ledd (Temporo-mandibular skjøt) og mer sjelden det Albue ledd. Dokumentasjonen for overmobilitet i leddene (hypermobilitet) utføres av Beightons poengsumsom bekrefter hypermobilitet på 5 av 9 mulige punkter.
Andre symptomer på leddene er generelle leddproblemer, kroniske nakkesmerter, bevege seg- og Hoftsmerter, Felles og Muskelsmertersom er vanskelige å behandle. Noen ganger peker smerte ("Tender ponits"), som er definert som et område som reagerer smertefullt på trykkbelastninger på 4 kg eller mindre. I tillegg er det økt risiko for brudd på grunn av redusert beinmasse kombinert med en unormal beinstruktur.
På grunn av skjørheten i bindevevet i blodkarene, er det en uttalt tendens til hematomerspontant eller som et resultat av traumer,
hovedsakelig i områder med fare for skade. Dette blir fulgt av en typisk i de berørte områdene brun pigmentering.
Etter skader observerer man en tendens til langvarig blødning med normale koagulasjonsverdier. Skjørheten i større blodkar kan utløses av anstrengelse, ulykker, svangerskap eller fødsel føre til massiv, livstruende blødning.
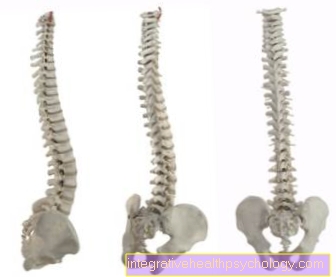
Siden andre bindevevsstrukturer også er dårligere, kan det bli det også viscera (Brokk / lyskebrokk), krumning av ryggraden (skoliose), Sprekker (brudd) av tarmene og livmor (Livmor), sekker (aneurisme) av blodkar og innsnevring av lungene fra fri luft i brystet (pneumothorax) kom.
I sjeldne tilfeller er øyeendringer assosiert med EDS som Astigmatisme (astigmatisme) eller grønn stjerne (glaukom) å observere.
diagnose
Diagnosen stilles på grunnlag av det kliniske utseendet, symptomene, og suppleres av en familieundersøkelse (familiehistorie). I tillegg kommer a Hudbiopsi hvor det fjernede hudvevet undersøkes ved bruk av et elektronmikroskop og dets kollagenstruktur blir vurdert. Differensieringen i de forskjellige typene av Ehlers-Danlos syndrom foregår ved hjelp av sekvensanalyse av DNA.
Klassifisering / typer
Type I, II: Klassisk fyr; Arv: autosomalt dominerende; Hoved symptomer: Hyperelastisitet og skjørhet i huden, atrofisk arrdannelse, ledens hypermobilitet; Årsak: kollagen V-formasjonsforstyrrelse
Type III: Hypermobile type; Arv: autosomalt dominerende; Hoved symptomer: generalisert leddhypermobilitet, hudinvolvering (hyperelastisitet og / eller myk, sårbar hud); Årsak: kollagen V-formasjonsforstyrrelse
Les mer om denne viktige typen på: Ehlers-Danlos syndrom type III
Type IV: Vaskulær type; Arv: autosomalt dominerende; Hoved symptomer: tynn gjennomskinnelig hud, rupturer i arteriene, tarmer og livmor, uttalt tendens til hematom; Årsak: kollagen III-formasjonsforstyrrelse
Type V: tilsvarer type I.
Type VI: Kyphoscoliotic type; Arv: autosomal recessiv; Hovedsymptomer: redusert spenning av muskulatur allerede ved fødselen ("Floppy baby"), forsinket utvikling av holder og støtte reflekser, lateral bøyning av Ryggrad (Skoliose), Opprinnelig årsak: Lsysl hydroksylase mangel
Type VII A / B: Arthrochalastic type; Arv: autosomalt dominerende; Hoved symptomer: alvorlig generalisert hypermobilitet i leddene med gjentatte dislokasjoner, medfødt bilateral hoftedlokasjon; Opprinnelig årsak: Type I kollagenlidelse
Type VII C: Dermatosparaktisk type; Arv: autosomalt dominerende; Hoved symptomer: uttalt hudens skjørhet, slapp hud, Opprinnelig årsak: Mangel på N-terminal procollagen I peptidase
Terapi og profylakse
Ingen årsaks fortsatt en symptomatisk terapi er for øyeblikket mulig, så profylakse for følgeskader er i forgrunnen. Skader og større belastning på leddene bør unngås. Så burde det være sikkert sportsom er assosiert med økt risiko for skader, utøves ikke. På grunn av den økte risikoen for komplikasjoner svangerskap og fødsel både Type I, II, IV og VI nøye overvåking er nødvendig.
Det skal også brukes med forkjølelse hostundertrykkende terapi og generelt til Regulering av avføringskonsistens bli respektert, fordi noe slikt Kolon ruptur (Tykktarmsbrudd) og a pneumothorax kan unngås. Gjennom tidlig fysioterapi, spesielt hos barn, kan de overtrekkbare leddene stabiliseres, noe som fører til lindring av symptomene i hele muskel-skjelettsystemet.
sår spesiell forsiktighet må utvises, og operasjoner skal bare utføres i nødsituasjoner fordi sårheling tar 3 til 4 ganger saktere enn vanlig.
prognose
Pasient med Ehlers-Danlos syndrom har vanligvis en normal forventet levealder. Imidlertid er sykdommen progressiv, så den fører til en stadig økende forverring av helsen. Hudsår og leddledninger påvirker pasientens livskvalitet, mens brudd på de store karene kan være livstruende.
Forventet levealder
Ehlers-Danlos syndrom er en kronisk sykdom som det fremdeles ikke er årsaksmessig behandling og derfor ingen kur. Dette betyr at det, med tanke på dagens medisinsk teknologi, ikke er mulig å gjøre noe med årsakene til Ehlers-Danlos syndrom og å kurere det fullstendig. Dessverre er man fremdeles ikke i stand til å bekjempe og behandle symptomene som oppstår. Den aktuelle pasienten kan bare oppfordres til alltid å være oppmerksom i hverdagen for ikke å legge for mye belastning på leddene og for å unngå skader på huden om mulig. Kirurgiske inngrep bør bare utføres i en nødsituasjon og hvis det ikke er tilstrekkelige alternativer.
I de fleste tilfeller er det progressivt med en økende forverring av symptomer og svekkelser i pasientens hverdag. Avhengig av type sykdom, har sykdommen forskjellige effekter på livene til de berørte. Endringene i leddene fører noen ganger til slitasjegikt og leddgikt i tidlig barndom, slik at barna senere lærer å gå og føttene kan bli deformerte. Med den økte risikoen for netthinneavløsning eller netthinneblødning blir synet også svekket.
Hvor sterkt symptomene til syvende og sist uttales hos hvert individ avhenger sterkt av typen Ehlers-Danlos-syndrom, og de kan også variere sterkt innenfor de enkelte undertypene. For de fleste typer Ehlers-Danlos syndrom er forventet levealder normal. Hos pasienter med Ehlers-Danlos syndrom type IV, som påvirker karene, reduseres forventet levealder betydelig på grunn av alvorlige komplikasjoner, for eksempel risikoen for spontan ruptur av en arterie, spesielt hovedpulsåren (med. Aortarivning) eller tykktarmen.
Det er rundt 37 år for kvinner og 34 år for menn.
En redusert forventet levealder kan også antas med Ehlers-Danlos syndrom type VI.









.jpg)