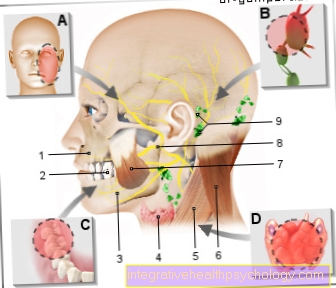
Genetiske sykdommer
definisjon
En genetisk sykdom eller arvelig sykdom er en sykdom som er forårsaket av en eller flere gener fra personen det gjelder. DNAet fungerer her som en direkte trigger av sykdommen. For de fleste genetiske sykdommer er de forårsakende genplasseringene kjent. Hvis man mistenker en genetisk sykdom, kan den respektive diagnosen derfor stilles gjennom en genetisk undersøkelse.
På den annen side er det også et stort antall sykdommer hvis forekomst har genetisk innflytelse eller blir diskutert, for eksempel diabetes mellitus ("diabetes"), osteoporose eller depresjon. Dette er såkalte disposisjoner, dvs. økt sannsynlighet for visse sykdommer. Disposisjoner skal skilles fra arvelige sykdommer.

Dette er vanlige arvelige sykdommer
I absolutte termer er arvelige sykdommer ikke vanlige, men de arvelige sykdommene som er oppført her forekommer ofte i sammenligning med andre genetiske sykdommer.
-
Marfans syndrom
-
Sikkelcelleanemi
-
Hemofili (hemofili A eller B)
-
Faktor V Leiden-mutasjon og resulterende APC-resistens
-
Rødgrønn svakhet
-
Glukose-6-fosfatdehydrogenase mangel (G6PD mangel)
-
Polydactyly ("flere fingre", også mulig som symptom ved andre sykdommer)
-
Trisomi 21 (Downs syndrom)
-
Chorea huntington
fører til
Arvelige sykdommer er ekstremt forskjellige i utseendet. De har i utgangspunktet bare en ting til felles: Årsaken til hver enkelt av dem ligger i DNA, dvs. i arvestoffet til personen det gjelder. Ulike forandringer kan forekomme her, for eksempel mutasjoner (utveksling av DNA-informasjon) eller sletting (mangel på bestemt genetisk materiale).
En stor mengde informasjon er kodet i arvematerialet, for eksempel “blåkopier” for forskjellige komponenter som er viktige for at en kroppscell fungerer. Dette kan for eksempel være enzymer, elektrolyttkanaler eller messenger-stoffer. Disse minste elementene leses da feil eller ikke i det hele tatt fra DNA, som da mangler i kroppens sofistikerte system. Feil eller manglende genetisk informasjon forårsaker derfor visse funksjonsfeil i kroppen. Disse forårsaker da symptomer i henhold til det funksjonelle systemet der ett element nå mangler.
Finn ut alt om emnet her: Den genetiske testen.
Slik blir arvelige sykdommer arvet
Hver arvelig sykdom blir arvet enten monogenetisk eller polygenetisk: Dette betyr at det er en eller flere genetiske lokasjoner som må endres for å føre til en sykdom.
Videre kan genetiske egenskaper alltid arves på en dominerende eller recessiv måte: Resessiv betyr at det må være en predisposisjon for denne spesielle arvelige sykdommen i både faderlig og morsgener. Ved dominerende arv er en endring (dvs. en forelder) nok til å utløse sykdommen. Det følger at med dominante arvelige sykdommer, vil også menneskene som er bærere bli syke - mens det med en recessiv arv vanligvis ikke engang er kjent at en tilsvarende genetisk disposisjon er til stede.
Det er også sykdommer som blir arvet gjennom kjønnskromosomene, for eksempel hemofili eller rødgrønn blindhet. Fasilitetene for dette er vanligvis på X-kromosomet, siden Y-kromosomet generelt er veldig lite og generelt kan lagre lite genetisk informasjon. Man snakker derfor om X-koblede arvelige sykdommer. Disse rammer vanligvis betydelig flere menn enn kvinner, da kvinner kan kompensere for all uriktig informasjon om X-kromosomet med det andre.
Hvordan en genetisk sykdom blir arvet, er vanligvis lett å undersøke hvis du er interessert.
Tester før fødsel
I prinsippet kan barnets genetiske materiale allerede undersøkes i livmoren for alle arvelige sykdommer hvis årsaksgenetiske lokaliteter er kjent. Imidlertid er genetiske analyser tidkrevende, så vanligvis analyseres bare den mistenkte genlokaliseringen - for dette må det igjen være en berettiget mistanke om en genetisk sykdom.
For en slik undersøkelse kan genetisk materiale tas fra fostervannet eller morkaken og brukes til analysen.
Imidlertid må det alltid huskes at enhver invasiv diagnose også innebærer en risiko for det ufødte barns liv. Slike punkteringer må derfor veies individuelt i hvert tilfelle.
Det finnes også målinger som kan indikere en genetisk sykdom, for eksempel måling av nuchal gjennomsiktighet som et tegn på trisomi 21. Slike metoder er ikke farlige for det ufødte barnet, men kan ikke tilby absolutt sikkerhet for at en genetisk sykdom er til stede. Så også her må en operasjon vurderes nøye.
Trisomi 21
Årsaken til trisomi 21 er kromosom 21, som ikke er til stede to ganger, men tre ganger hos berørte personer. Denne varianten av DNA skapes når kromosomene er fordelt i foreldrenes kimceller, dvs. sædcellene eller eggcellene. Det er derfor en "distribusjonsfeil" og ikke en endring i det faktiske arvestoffet. Dette forklarer hvorfor trisomi 21 kan oppstå spontant i hver familie, og hvorfor sannsynligheten for å få et barn med Downs syndrom er den samme i alle familier. Strengt tatt bør trisomi 21 - som andre trisomier - ikke regnes som en arvelig sykdom i sann forstand. Likevel er trisomi 21 den vanligste DNA-relaterte sykdommen hos nyfødte.
Egenskaper ved det endrede settet med kromosomer i Downs syndrom kan allerede sees hos det ufødte barnet i livmoren: Vekstforsinkelser og defekter kan føre til blant annet en hodeskalle som er for liten, korte bein i lår og overarm og hjertefeil. En stor mengde fostervann kan også være en indikasjon på trisomi 21, siden berørte ufødte barn drikker eller svelger relativt lite fostervann. Ingen av disse funksjonene er imidlertid definitive tegn på Downs syndrom!
I tillegg til de nevnte tegn på veksthemming, viser barn med Downs syndrom ofte også forsinket utvikling, for eksempel innen språk og motorikk. Personer som er rammet av Downs syndrom viser ofte bemerkelsesverdige sosiale ferdigheter, mens intelligens ofte forblir under gjennomsnittet. Imidlertid er berørte mennesker veldig forskjellige i disse egenskapene, og det er ikke uvanlig at de oppgraderer seg fra skolen etter å ha fått god støtte.
Senere i livet har personer med trisomi 21 økt risiko for å få diagnosen visse sykdommer. Disse inkluderer Alzheimers sykdom, epilepsi og kreft, spesielt leukemi. Likevel fortsetter levealderen til personer med Downs syndrom å øke: I mellomtiden når berørte mennesker ofte 60 eller 70 år.
Du kan finne mer informasjon på vår hjemmeside Downs syndrom
Alpha-1 antitrypsinmangel
Alpha-1 antitrypsinmangel kan ha forskjellige former og former, avhengig av de nøyaktige genetiske egenskapene til personen som er berørt. Dette betyr at ikke alle alfa-1 antitrypsinmangel fører til symptomer. I det følgende vil bare den klinisk iøynefallende typen (PiZZ) av denne genetisk bestemte sykdommen bli diskutert.
Enzymdefekten som er tilstede i denne sykdommen forårsaker sammenbrudd og ombygging av byggesteiner i organvevet hos berørte personer. I tillegg filtreres de mangelfulle proteinene ut av blodet av leveren og akkumuleres der. Dette kan føre til betennelse i leveren (hepatitt), skrumplever eller leverkreft. Luftveiene i lungene blir ustabile på grunn av mangel på stabilt vev, og de kollapser raskere: Det kliniske bildet av KOLS (kronisk obstruktiv lungesykdom) utvikler seg. Dette kliniske bildet er ofte det første symptomet på alfa-1 antitrypsinmangel, så enhver person med KOLS i yngre alder bør sjekkes for alfa-1 antitrypsinmangel.
Hvis sykdommen har vedvart i lang tid, kan lungene overoppblåse, da luften du puster ikke kan pustes ut ordentlig gjennom de ustabile luftveiene og samler seg i lungene. Som en terapi, i tillegg til konsekvent å unngå sigarettrøyking og regelmessige vaksiner for å forhindre luftveissykdommer, bør det også iverksettes medisinske tiltak: Det manglende alpha-1-antitrypsin kan administreres intravenøst for å lindre symptomene så langt det er mulig og stoppe sykdomsforløpet.
Du kan finne mer informasjon på vår hjemmeside Alpha-1 antitrypsinmangel
hemofili
Gruppen av hemofili er kjent som “hemophilia”, da dette uttrykket beskriver hovedsymptomet på denne arvelige sykdommen veldig presist: de berørte blør lenger og avhengig av alvorlighetsgraden av sykdommen, oftere enn upåvirket.
Blødning stoppes vanligvis av det som er kjent som koagulasjonskaskaden, en endogen signalvei som forhindrer overdreven blodtap. I dette koagulasjonssystemet spiller 13 faktorer en rolle, som aktiverer hverandre etter hverandre. Dette kan tenkes som en serie med dominoer: hvis du treffer en stein (koagulasjonsfaktor), aktiveres den neste, og så videre. På slutten av denne signalveien eller dominoene er det blodkoagulasjon. Med hemofili mangler en viss faktor - avhengig av den spesifikke undertypen til sykdommen: kjedereaksjonen bryter av her.
Terapi for sykdommen kan utføres ved å bestemme den manglende faktoren og legge den utenfra. Berørte mennesker må derfor regelmessig injisere seg et preparat med denne koagulasjonsfaktoren slik at resten av kjedereaksjonen kan finne sted.
Du kan finne mer informasjon på vår hjemmeside Blodsykdom
Cystisk fibrose
I den genetiske sykdommen cystisk fibrose - også kjent som cystisk fibrose - er det en feilproduksjon av ionekanaler, mer presist av kloridkanaler. Som et resultat endres sammensetningen av kroppssekretene (f.eks. Svette, sekreter fra luftveiene og bukspyttkjertelen) hos de berørte personer: Siden mangelen på klorid betyr at mindre vann trekkes inn i kanalen til den respektive kjertelen, er sekresjonen relativt tyktflytende.
Som et resultat utvikles det vanligvis symptomer i fordøyelseskanalen, da sekresjonen med fordøyelsesenzymer ikke kan strømme godt fra bukspyttkjertelen inn i tarmen og dermed skader selve bukspyttkjertelen. I tillegg er fordøyelsessykdommer som fet avføring, diaré og den resulterende lave kroppsvekten vanlige.
Den andre store gruppen av symptomer utvikler seg vanligvis i lungene: Siden slimet som naturlig forekommer i lungene er mer tyktflytende enn hos friske mennesker, er det vanskeligere å fjerne det fra cilia. Dette kan føre til kronisk hoste og blokkering av bronkiene (bronkieektase). Den større mengden lungesekresjon gir også et godt miljø for vekst av bakterier, noe som resulterer i hyppige luftveisinfeksjoner og lungebetennelse.
Cystisk fibrose behandles symptomatisk med slimløsende midler, fordøyelsesenzymer og antibiotika for infeksjoner.
Du finner mer om dette på vår hjemmeside Cystisk fibrose
Faktor V Leiden og APC-motstand
En faktor V Leiden-mutasjon innebærer en endring i genetisk informasjon som kan forårsake økt blodpropp. Årsaken til dette er faktor V i kroppens såkalte koaguleringskaskade: denne signalveien sikrer at i tilfelle en skade blir såret lukket av kroppens egne "klebeproteiner" (fibrin). Det er 13 faktorer i denne signalveien, som er navngitt med romertall (det betyr “Faktor 5 som lider”!). Faktoren V har en gunstig effekt på dannelsen av en fibrinplugg, men kan også hemmes av det såkalte aktiverte protein C (APC for kort). Dette spiller en viktig rolle i reguleringen av denne signalveien og i å forhindre overdreven blodpropp.
Den muterte faktoren V er til stede hos de berørte individer, men svarer ikke til APC. Kroppen mangler en viktig "sikkerhetsinnretning" på dette tidspunktet for å forhindre blodpropp uten grunn, noe som til og med kan blokkere kar og derved forårsake sirkulasjonsforstyrrelser.
Statistisk sett er det mer sannsynlig at personer som er påvirket av en faktor V Leiden-mutasjon, opplever en trombotisk hendelse (dvs. trombose eller lungeemboli), selv uten en historie med typiske risikofaktorer. Rent teknisk snakker man også om “trombofili”, det vil si en tendens til koagulering.
Du finner mer om dette på vår hjemmeside Faktor V Leiden
Gauchersykdom
Ved Gauchers sykdom forårsaker endringen i DNA-informasjon en defekt i et enzym som er involvert i lipidmetabolisme, mer presist glukoserebrosidase: dette hjelper til med å bryte ned gamle cellulære komponenter. I tilfelle en mangel kan det være en reduksjon i funksjonalitet eller til og med tap av funksjonalitet, og følgelig vises symptomene i barndommen eller i ung voksen alder.
Symptomene på Gaucher sykdom skyldes i stor grad en utvidelse av leveren og milten, hvor veksten kroppen prøver å kompensere for mangelen på enzymer. Dette øker nedbrytningen av alle blodkomponenter, som kan gjenkjennes i blodtellingen og brukes som en diagnostisk indikator sammen med den forstørrede leveren og milten.
Den manglende enzymet glukocerebrosidase kan brukes terapeutisk som et medikament. Prognosen og forløpet av Gauchers sykdom avhenger i stor grad av alvorlighetsgraden av enzymets funksjonstap.
For mer informasjon, les videre her: Gauchers sykdom.
Oslers sykdom
Oslers sykdom er en arvelig sykdom som er preget av en sterk vasodilatasjon. I prinsippet kan denne utvidelsen av karene skje hvor som helst, både på huden og på indre organer. Veggene på de forstørrede karene er relativt tynne og rives lett. Som et resultat blør de berørte områdene raskt.
Vasodilatasjon i ansiktet og neseslimhinnen forekommer spesielt ofte, slik at de berørte vanligvis klager over hyppige neseblod og små flekkete blødninger i ansiktet.
Hvis man mistenker Oslers sykdom, bør passende diagnostikk utføres, siden vasodilatasjonen også kan forekomme i vitale organer eller organer med god blodtilførsel, for eksempel lungene, hjernen eller leveren, der blødning fra et ødelagt kar er farlig.
Du kan finne mer om dette emnet på vår hjemmeside Oslers sykdom
Recklinghausen sykdom
Neurofibromatosis type 1 - eller Recklinghausens sykdom - er en genetisk sykdom der de berørte ofte utvikler svulster på nervene som dekker nervene. Svulstene som utvikler seg kan være både godartede og ondartede og vises i ung alder.
Typiske svulster er imidlertid godartede neurofibromer: Disse består av celler som kapper og isolerer nerven som en elektrisk kabel, så vel som det omkringliggende bindevevet. De er godartede, dvs. ikke-spredte og langsomt voksende svulster.
Imidlertid kan kirurgi for å fjerne neurofibromer være vanskelig, siden de ofte er godt festet til nerven og den tilsvarende nerven da må fjernes. Ikke desto mindre er dette det eneste behandlingsalternativet for symptomatisk nevrofibrom, da kausal terapi for denne arvelige sykdommen ikke er mulig.
Du kan finne mer om dette emnet på vår hjemmeside Neurofibromatosis type 1
Muskeldystrofi
Begrepet muskeldystrofi beskriver en gruppe arvelige sykdommer der visse muskelkomponenter ikke kan eller ikke kan samles riktig av kroppens celler. Som et resultat utvikler de berørte vanligvis muskelsvakhet allerede i barndommen og i ungdomstiden, og dette kan resultere i tap av muskelmasse, bevegelsesbegrensninger og til og med fysiske funksjonshemninger.
Hvis det er mistanke om tilstedeværelse av muskeldystrofi, bør blodverdiene først bestemmes. Hvis verdiene samsvarer med den mistenkte diagnosen, kan det fortsatt utføres en muskelbiopsi: en liten vevsprøve tas fra muskelen, som deretter undersøkes mikroskopisk for celledefekter. En genetisk undersøkelse er også mulig å etablere diagnosen, siden de tilsvarende genetiske lokaliseringene vanligvis er kjent for de forskjellige former for muskeldystrofi og måtte endres. En kausal terapi for muskeldystrofier er ikke kjent.
Du kan finne mer om dette emnet på vår hjemmeside Muskeldystrofi
Xeroderma pigmentosum
Xeroderma pigmentosum er en sjelden arvelig sykdom der visse enzymer i huden til den berørte ikke fungerer. Disse enzymene tar normalt vare på reparasjonen i DNAet, som kan bli skadet av sollys eller UVB-lyset. UVB-skaden kan forårsake hudkreft hos berørte mennesker så vel som hos alle andre mennesker, men med Xeroderma Pigmentosum blir prosessen fremskyndet av mangelen på reparasjonsmekanismer. Som et resultat utvikler de berørte menneskene alvorlige former for hudkreft i barne- og ungdomsårene og etter en kort eksponering for sollys.
En kausal terapi er ennå ikke mulig. De berørte menneskene må unngå sollys for livet, og det er grunnen til kallenavnet "måneskinnbarn" som har etablert seg for de berørte (noen ganger veldig unge) berørte. I tillegg bør disse menneskene overvåkes av en hudlege for regelmessig screening av hudkreft for å fjerne nyutviklet hudkreft umiddelbart. Hvis disse tiltakene følges strengt, er forventet levealder for en person med xeroderma pigmentosum omtrent den samme som for en upåvirket person.
Du kan finne mer om denne sykdommen på nettstedet vårt Xeroderma pigmentosum
Lynch syndrom
Lynch syndrom er en endring i DNA som forårsaker et mangelfullt enzym i kroppens celler.Hos de berørte er en viss mekanisme mangelfull, noe som ellers er ment å beskytte cellene mot degenerasjon, dvs. ukontrollert vekst - personer med Lynch syndrom har derfor en sterkt økt risiko for å utvikle kreft.
Tykktarmskreft forekommer ofte fordi cellene naturlig ofte deler seg her uansett og feil i programmering av vekst og død av en celle blir tydeligere tydelig. Berørte mennesker utvikler ofte en svulst i tykktarmen i en uvanlig ung alder, dvs. før fylte 50 år, som da kalles HNPCC (arvelig ikke-polypøs tykktarmskreft). Imidlertid vil ikke alle som har genetisk sammensetning av Lynch syndrom utvikle tykktarmskreft. På den annen side kan andre organer også utvikle en svulst, siden de genetiske predisposisjonene som favoriserer utviklingen av en svulst, er til stede i alle kroppens celler. Regelmessige kontroller og forebyggende undersøkelser er derfor nødvendige for de som er rammet av Lynch syndrom for å kunne behandle svulster som utvikler seg på et tidlig tidspunkt.
Du kan finne mer om dette emnet på vår hjemmeside Lynch syndrom